Polycystic Kidney Disease: Understanding the Genetics and How It's Managed
Polycystic kidney disease (PKD) isn't just about fluid-filled sacs in the kidneys. It's a genetic time bomb that quietly damages organs over decades, often without symptoms until it's too late. About 600,000 people in the U.S. live with it, and half of those with the most common form will need dialysis or a transplant by age 60. What makes PKD so dangerous isn't just the cysts - it's how little most people know about it until they're already deep into kidney failure.
What Exactly Is Polycystic Kidney Disease?
PDK is a hereditary condition where hundreds or even thousands of cysts grow inside the kidneys. These aren't harmless lumps. They expand over time, pushing out healthy tissue, squeezing blood vessels, and slowly turning functional kidney cells into scar tissue. By the time someone reaches their 50s or 60s, their kidneys can weigh up to 30 pounds - that’s heavier than a large dog - and still barely function.
There are two main types, and they’re as different as night and day. The most common form, autosomal dominant polycystic kidney disease (ADPKD), affects more than 98% of cases. It’s passed down from just one parent who has the mutated gene. The rarer form, autosomal recessive polycystic kidney disease (ARPKD), requires both parents to carry the faulty gene. Even then, each child only has a 25% chance of developing it. ARPKD usually shows up in newborns or toddlers, while ADPKD often stays silent until adulthood.
The Genes Behind the Cysts
It all comes down to three genes: PKD1, PKD2, and PKHD1. Mutations in PKD1 cause about 78% of ADPKD cases and tend to be the most aggressive. PKD2 mutations account for 15% and usually mean slower progression. People with PKD1 often reach kidney failure 20 years earlier than those with PKD2. Then there’s PKHD1, linked to ARPKD. This gene doesn’t just affect the kidneys - it also messes with the liver, leading to bile duct problems and scarring.
Here’s the twist: even if you inherit the faulty gene, your disease doesn’t follow a predictable path. Two siblings with the exact same mutation can have wildly different outcomes. One might need a transplant at 45, while the other holds off until 70. Why? Scientists think other genes, lifestyle factors, and even random cellular errors play a role. That’s why doctors can’t just say, “You’ll fail in 15 years.” They can only watch, test, and react.
ADPKD vs. ARPKD: Key Differences
| Feature | ADPKD | ARPKD |
|---|---|---|
| Prevalence | 1 in 400-1,000 people | 1 in 20,000 people |
| Onset | 30-40 years old (often unnoticed until then) | Infancy or early childhood |
| Genetic Cause | One mutated copy of PKD1 or PKD2 | Two mutated copies of PKHD1 |
| Family History | 90% inherited; 10% new mutation | Usually no family history |
| Major Complications | Hypertension, kidney failure, liver cysts | Breathing problems at birth, liver fibrosis, kidney failure |
| Progression to Kidney Failure | 50% by age 60 | Often by teens or early adulthood |
ADPKD sneaks up on you. Many people live for years with high blood pressure, back pain, or frequent urinary infections - and never connect them to PKD. It’s only when a scan shows dozens of cysts that the diagnosis clicks. ARPKD is harder to ignore. Babies born with it often struggle to breathe because their kidneys are too swollen to function. Many don’t survive the first month. Those who do face lifelong liver damage and kidney problems.
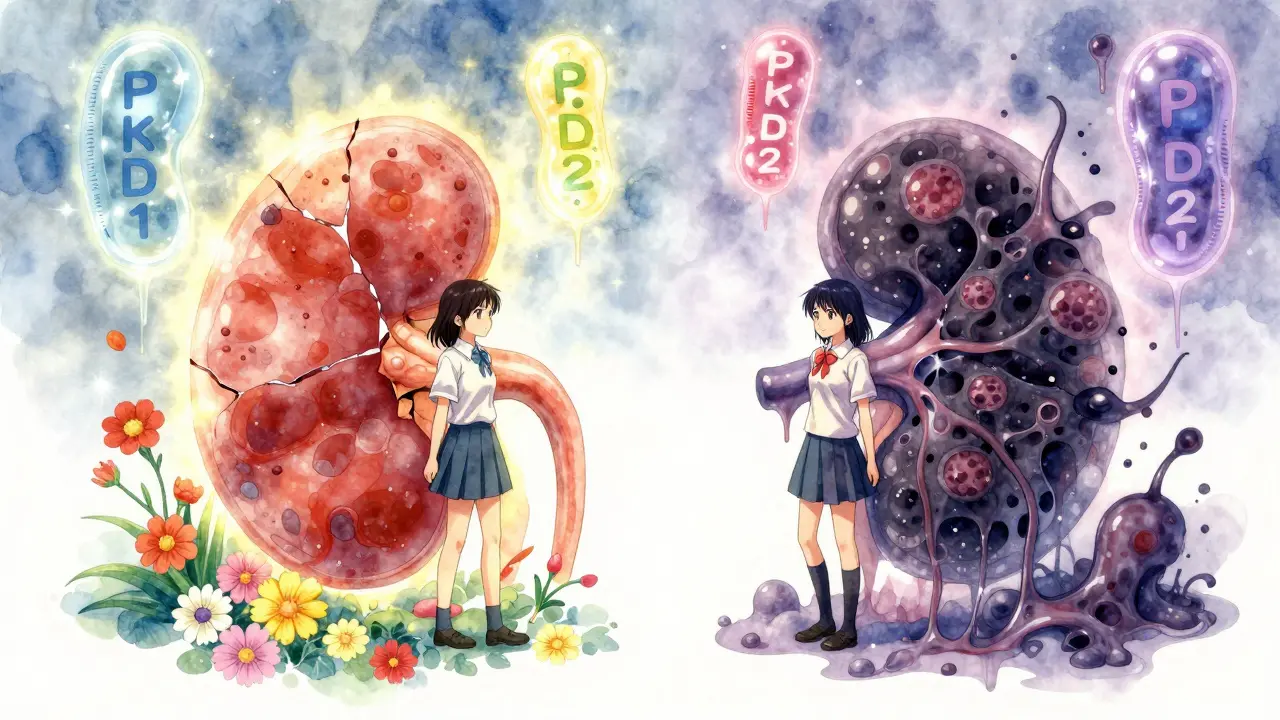
How Is It Diagnosed?
If you have a family history of PKD, doctors don’t wait for symptoms. They use imaging. A simple ultrasound can spot cysts in people over 30. For younger patients with a family history, a CT scan or MRI gives a clearer picture. The rule of thumb? If you’re 30-39 with a parent who has ADPKD and have at least 10 cysts in one kidney, you have it.
Genetic testing is now an option. A blood test can check for mutations in PKD1, PKD2, or PKHD1. It costs around $1,200 and is covered by many insurers if you have a family history or early symptoms. It’s especially useful for people planning to have kids - if you carry the gene, your child has a 50% chance of inheriting ADPKD.
Managing the Disease: Beyond Waiting
There’s no cure. But that doesn’t mean you do nothing. The goal is to delay kidney failure as long as possible - and improve quality of life while you wait.
- Control your blood pressure. This is the single most important thing. Keeping it below 130/80 mmHg can slow cyst growth. Many patients start on ACE inhibitors or ARBs - drugs that protect the kidneys while lowering pressure.
- Monitor kidney function. Your eGFR (estimated glomerular filtration rate) should be checked at least once a year. If it drops below 60 mL/min/1.73m², testing should go quarterly.
- Use tolvaptan (Jynarque). This is the only FDA-approved drug that directly targets ADPKD progression. It blocks a hormone that makes cysts grow. In clinical trials, it slowed kidney decline by 1.3 mL/min/year. But it’s expensive - over $115,000 a year - and only for patients with rapidly progressing disease.
- Watch your diet. Limit salt, avoid excessive protein, and stay hydrated. Alcohol and caffeine can worsen symptoms. Some studies suggest low-sodium, plant-rich diets help preserve kidney function longer.
- Manage pain. Cysts can burst, bleed, or get infected. Chronic pain affects 78% of patients. Over-the-counter painkillers like acetaminophen are safe. Avoid NSAIDs like ibuprofen - they hurt the kidneys.
The Emotional Toll
Living with PKD isn’t just physical. A 2022 survey by the PKD Foundation found that 63% of patients struggle with anxiety about kidney failure. One Reddit user wrote: “It took seven years and three doctors before anyone believed me - even though my dad died of PKD at 52.”
Many feel guilty if they pass the gene to their kids. Others feel invisible because the disease doesn’t show outward signs until it’s advanced. Support groups, genetic counseling, and mental health care aren’t optional extras - they’re part of treatment.

What’s Coming Next?
Research is moving fast. The drug tolvaptan was approved in 2018, but now there are at least five new drugs in phase 3 trials. Lixivaptan, expected to be reviewed in 2024, looks promising. Bardoxolone methyl showed improved kidney function in early trials. These aren’t cures - but they’re steps toward slowing the disease more effectively.
Gene editing tools like CRISPR are being tested in labs. While still years away from clinical use, they offer real hope for one day correcting the faulty gene before cysts even form.
When It’s Time for Dialysis or Transplant
By age 70, 75% of ADPKD patients will need dialysis or a transplant. The good news? Transplant success rates for PKD patients are among the highest in kidney disease - over 90% at five years. The bad news? Wait times vary. In some regions, you might wait three years. In others, five. Blood type matters. O-negative patients often wait the longest.
Many patients choose to get on the transplant list early - even if their kidneys still work. It gives them time to get healthy, find a donor, and plan ahead. Living donors are common: a sibling, parent, or even a friend can donate one kidney safely.
Can you prevent polycystic kidney disease if it runs in your family?
No, you can’t prevent inheriting the gene. But you can prevent or delay complications. Genetic testing can confirm if you carry the mutation. If you do, early blood pressure control, regular monitoring, and avoiding kidney stressors (like NSAIDs, dehydration, or high-salt diets) can delay kidney failure by decades. For couples planning children, preimplantation genetic testing (PGT) during IVF can screen embryos for the mutation.
Does having PKD mean you’ll definitely need a kidney transplant?
Not necessarily. About half of people with ADPKD reach kidney failure by age 60. But many live into their 70s or 80s with functioning kidneys - especially if they control their blood pressure tightly, avoid smoking, and follow medical advice. Some never need dialysis. Transplant isn’t the only outcome - it’s one possible outcome, and often a last resort.
Can children be tested for ADPKD before symptoms appear?
Yes, but it’s rarely recommended. Most guidelines advise against testing kids under 18 unless symptoms appear. Why? Because there’s no treatment for children with early-stage ADPKD, and knowing the diagnosis early can cause anxiety, affect insurance, or impact self-image. If a child has high blood pressure or pain, then testing makes sense. Otherwise, waiting until adulthood is standard practice.
Is PKD only a kidney problem?
No. ADPKD affects other organs too. Cysts can form in the liver, pancreas, and even the brain (causing aneurysms in 10% of patients). Heart valves can leak. Hernias are common. ARPKD primarily affects the liver, causing severe scarring and bile duct issues. That’s why PKD patients need more than just kidney care - they need liver scans, brain imaging, and heart checks as part of routine monitoring.
Why is PKD so expensive to treat?
Because patients are younger. Most kidney failure patients are over 65 and on Medicare. PKD patients often develop end-stage disease in their 40s or 50s - meaning they’re on dialysis for 20+ years. A single year of dialysis costs $90,000. A transplant costs $400,000 upfront. Add in medications like tolvaptan ($115,000/year), frequent imaging, and specialist visits, and the lifetime cost can exceed $2 million. That’s why PKD accounts for 5% of Medicare’s ESRD budget despite only causing 2% of cases.
What Should You Do Now?
If you have a family history of PKD, get tested. Don’t wait for symptoms. If you’ve been diagnosed, start managing blood pressure now - even if it’s only slightly high. Talk to a nephrologist. Join a patient support group. Know your numbers: eGFR, blood pressure, cyst size. And if you’re thinking about having kids, talk to a genetic counselor.
PKD is not a death sentence. It’s a chronic condition - one that can be managed, delayed, and lived with. The difference between thriving and surviving? Early action. Knowledge. And not giving up on the future.
Jazminn Jones
March 9, 2026 AT 09:44It is profoundly concerning that the medical establishment continues to treat polycystic kidney disease as a monolithic entity when the phenotypic variance-particularly between PKD1 and PKD2-is so statistically significant. The notion that ‘watchful waiting’ constitutes a viable clinical strategy is, frankly, archaic. We must pivot toward stratified, genomics-informed intervention protocols, not reactive palliation. The current standard of care is a failure of translational medicine.
Stephen Rudd
March 10, 2026 AT 05:09Let me be blunt: this whole article reads like a pharmaceutical industry white paper. Tolvaptan costs over $100k a year? And you’re telling people this is the ‘only’ option? What about lifestyle interventions? The fact that you mention diet but bury it under jargon speaks volumes. This isn’t medicine-it’s a revenue model disguised as science.
Erica Santos
March 11, 2026 AT 05:02Oh wow, so we’ve officially turned human suffering into a spreadsheet? ‘By age 60, 50% need transplant’-how poetic. Meanwhile, the real tragedy is that we’ve normalized waiting for people to collapse before we bother to act. We don’t need more data. We need moral courage. And maybe a system that doesn’t treat kidneys like disposable widgets.
George Vou
March 11, 2026 AT 15:32ok so here's the thing... i read this whole thing and honestly? i think the whole 'genetic time bomb' thing is just fearmongering. my cousin has pkd and she's 58 and still runs marathons. they're just selling drugs. also, why do they keep saying 'cysts'? aren't they just water balloons? like... why make it sound so scary? also, i heard the government is putting tracking chips in dialysis machines. just saying.
Scott Easterling
March 12, 2026 AT 14:02Wait-hold on. Let me get this straight: you’re telling me that if I have one bad gene, I’m basically doomed? And that’s it? No second opinions? No alternatives? What about the 10% of cases with new mutations? Who’s to say those aren’t just misdiagnosed? And why is everyone so obsessed with ‘early detection’? Sounds like a surveillance state wrapped in a stethoscope.
Mantooth Lehto
March 13, 2026 AT 21:17I just lost my brother to this. I didn't know anything until it was too late. I'm so angry. I'm so tired. I just want someone to listen. Please. If you have this-please, please, please get checked. I don't want anyone else to feel this alone. 💔
Melba Miller
March 15, 2026 AT 13:04Why is it always Americans who act like PKD is some uniquely American tragedy? We have the same disease here. We have the same costs. We have the same silence. But you? You turn it into a narrative about healthcare failures. What about the global reality? People in India, Nigeria, Brazil-they don’t have access to ultrasounds, let alone tolvaptan. This isn’t a story about American medicine. It’s a story about global neglect.
Katy Shamitz
March 17, 2026 AT 02:45Thank you for writing this. I’ve been living with ADPKD for 12 years, and no one ever talks about the loneliness. The way your friends slowly stop asking if you’re ‘feeling okay’ because they don’t know how to respond. The guilt of wondering if your kids will inherit it. The fear that you’re a burden. This article didn’t just inform me-it made me feel seen. You’re not alone. I see you. I’m here.
Nicholas Gama
March 18, 2026 AT 21:13CRISPR won’t fix this. It’s a distraction. The real issue? The patent system. Tolvaptan’s patent expires in 2029. Until then, it’s a $115k racket. Gene therapy? Too expensive. Transplant? Too slow. The system isn’t broken-it’s designed this way. Profit over patients. Always.
Mary Beth Brook
March 20, 2026 AT 00:15ADPKD progression is modulated by mTOR and cAMP signaling cascades. Tolvaptan’s V2 receptor antagonism suppresses cystogenesis via aquaporin-2 downregulation. However, the heterogeneity in PKD1/PKD2 penetrance suggests epistatic interactions with modifier loci-likely on chr4q21 and chr16p13. Early intervention with SGLT2 inhibitors may offer synergistic renoprotection. Clinical trials are underway.
Neeti Rustagi
March 21, 2026 AT 10:08As a physician from India, I see PKD patients daily-many without access to imaging or genetic testing. The article is accurate in its science, yet incomplete in its context. We need low-cost ultrasound protocols, community health worker training, and subsidized generic alternatives. Knowledge is power, but access is justice. Let us not mistake data for equity.